nwalign
Globally align two sequences using Needleman-Wunsch algorithm
Syntax
Description
Score = nwalign(Seq1,Seq2)Seq1 and Seq2. The scale factor used to
calculate the score is provided by ScoringMatrix.
Score = nwalign(Seq1,Seq2,Name=Value)
Examples
Input Arguments
Name-Value Arguments
Specify optional pairs of arguments as
Name1=Value1,...,NameN=ValueN, where Name is
the argument name and Value is the corresponding value.
Name-value arguments must appear after other arguments, but the order of the
pairs does not matter.
Example: [s,a] =
nwalign("HEAGAWGHEE","PAWHEAE",GapOpen=5,ShowScore=true) specifies to use the
value of 5 as a penalty for gap opening and to show the scoring space and winning
path.
Before R2021a, use commas to separate each name and value, and enclose
Name in quotes.
Example: [s,a] =
nwalign("HEAGAWGHEE","PAWHEAE",'GapOpen',5,'ShowScore',true)
Type of sequence, specified as "AA" (amino acid) or
"NT" (nucleotide).
Data Types: char | string
Scoring matrix for the global alignment, specified as a character vector, string scalar, or numeric matrix.
You can specify a scoring matrix name. Valid choices are:
"BLOSUM50"(default for amino acid sequences)"NUC44"(default for nucleotide sequences)"BLOSUM62""BLOSUM30"increasing by5up to"BLOSUM90""BLOSUM100""PAM10"increasing by10up to"PAM500""DAYHOFF""GONNET"
Note
The above scoring matrices, provided with the software, also include a scale
factor that converts the units of the output score to bits. You can also specify
the Scale name-value argument to specify an additional scale
factor to convert the output score from bits to another unit.
You can also specify a numeric matrix, such as the one returned by the
blosum, pam, dayhoff,
gonnet, or nuc44 function.
Note
If you use a scoring matrix that you created or was created by one of these scoring matrix functions, the matrix does not include a scale factor. The output score will be returned in the same units as the scoring matrix. You can use the
Scalename-value argument to specify a scale factor to convert the output score to another unit.If you need to compile
nwaligninto a standalone application or software component using MATLAB® Compiler™, use a numeric matrix instead of the scoring matrix name.
Data Types: double | char | string
Scale factor applied to the output score, specified as a numeric scalar or vector.
If you specify a vector, the function returns Score as a vector
of the same length. By default, there is no scaling or change in the units of the
output score.
Use this argument to control the units of the output scores. For example, if the
output score is initially determined in bits, you can specify
Scale=log(2) to return the output score in nats instead.
Note
If the
ScoringMatrixargument also specifies a scale factor, then the function uses it first to scale the output score, then applies the scale factor specified by theScaleargument to rescale the output score.Before comparing alignment scores from multiple alignments, ensure that the scores are in the same units.
Data Types: double
Penalty for opening a gap, specified as a positive scalar.
Data Types: double
Penalty for extending a gap using the affine gap penalty scheme, specified as a positive scalar.
If you specify this value, the function uses the affine gap penalty scheme, that
is, it scores the first gap using the GapOpen value and scores
subsequent gaps using the ExtendGap value. If you do not specify
this value, the function scores all gaps equally, using the
GapOpen penalty.
Data Types: double
Flag to perform a semiglocal alignment, specified as a numeric or logical
1 (true) or 0
(false).
In a semiglobal alignment, gap penalties at the end of the sequences are null.
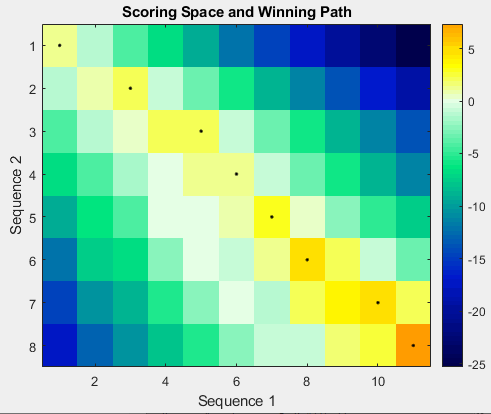
Flag to display the scoring space and winning path of the alignment, specified as
a numeric or logical 1 (true) or 0
(false).
The scoring space is a heat map displaying the best scores for all the partial
alignments of two sequences. The color of each (n1,n2) coordinate
in the scoring space represents the best score for the pairing of subsequences
Seq1(1:n1) and Seq2(1:n2), where
n1 is a position in Seq1 and
n2 is a position in Seq2. The best score for a
pairing of specific subsequences is determined by scoring all possible alignments of
the subsequences by summing matches and gap penalties.
The winning path is represented by black dots in the scoring space, and it
illustrates the pairing of positions in the optimal global alignment. The color of the
last point (lower right) of the winning path represents the optimal global alignment
score for the two sequences and is the Score output.
Note
The scoring space visually indicates if there are potential alternate winning paths, which is useful when aligning sequences with big gaps. Visual patterns in the scoring space can also indicate a possible sequence rearrangement.
Output Arguments
References
[1] Durbin, Richard, Sean R. Eddy, Anders Krogh, and Graeme Mitchison. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. 1st ed. Cambridge University Press, 1998.
Version History
Introduced before R2006a
See Also
aa2int | aminolookup | baselookup | blosum | dayhoff | gonnet | int2aa | int2nt | localalign | multialign | nt2aa | nt2int | nuc44 | pam | profalign | seqdotplot | swalign