kmedoids
k-medoids clustering
Syntax
Description
idx = kmedoids(X,k)X into k clusters, and returns an
n-by-1 vector idx containing cluster
indices of each observation. Rows of X correspond to points and
columns correspond to variables. By default, kmedoids uses
squared Euclidean distance metric and the k-means++
algorithm for choosing initial cluster medoid positions.
idx = kmedoids(X,k,Name,Value)Name,Value pair
arguments.
Examples
Randomly generate data.
rng('default'); % For reproducibility X = [randn(100,2)*0.75+ones(100,2); randn(100,2)*0.55-ones(100,2)]; figure; plot(X(:,1),X(:,2),'.'); title('Randomly Generated Data');
Group data into two clusters using kmedoids. Use the cityblock distance metric.
opts = statset('Display','iter'); [idx,C,sumd,d,midx,info] = kmedoids(X,2,'Distance','cityblock','Options',opts);
rep iter sum
1 1 209.856
1 2 209.856
Best total sum of distances = 209.856
info is a struct that contains information about how the algorithm was executed. For example, bestReplicate field indicates the replicate that was used to produce the final solution. In this example, the replicate number 1 was used since the default number of replicates is 1 for the default algorithm, which is pam in this case.
info
info = struct with fields:
algorithm: 'pam'
start: 'plus'
distance: 'cityblock'
iterations: 2
bestReplicate: 1
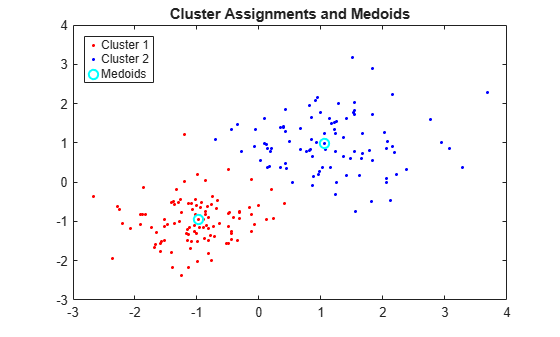
Plot the clusters and the cluster medoids.
figure; plot(X(idx==1,1),X(idx==1,2),'r.','MarkerSize',7) hold on plot(X(idx==2,1),X(idx==2,2),'b.','MarkerSize',7) plot(C(:,1),C(:,2),'co',... 'MarkerSize',7,'LineWidth',1.5) legend('Cluster 1','Cluster 2','Medoids',... 'Location','NW'); title('Cluster Assignments and Medoids'); hold off
This example uses "Mushroom" data set [3][4][5]
[6][7] from the UCI machine learning archive [7], described in https://archive.ics.uci.edu/dataset/73/mushroom. The data
set includes 22 predictors for 8,124 observations of various mushrooms. The
predictors are categorical data types. For example, cap shape is categorized
with features of 'b' for bell-shaped cap and
'c' for conical. Mushroom color is also categorized
with features of 'n' for brown, and
'p' for pink. The data set also includes a
classification for each mushroom of either edible or poisonous.
Since the features of the mushroom data set are categorical, it is not possible to define the mean of several data points, and therefore the widely-used k-means clustering algorithm cannot be meaningfully applied to this data set. k-medoids is a related algorithm that partitions data into k distinct clusters, by finding medoids that minimize the sum of dissimilarities between points in the data and their nearest medoid.
The medoid of a set is a member of that set whose average dissimilarity with the other members of the set is the smallest. Similarity can be defined for many types of data that do not allow a mean to be calculated, allowing k-medoids to be used for a broader range of problems than k-means.
Using k-medoids, this example clusters the mushrooms into two groups, based on the predictors provided. It then explores the relationship between those clusters and the classifications of the mushrooms as either edible or poisonous.
This example assumes that you have downloaded the "Mushroom" data set
[3][4][5]
[6][7] from the UCI database (https://archive.ics.uci.edu/dataset/73/mushroom) and
saved the text files agaricus-lepiota.data and agaricus-lepiota.names in
your current directory. There is no column header line in the data, so
readtable uses the default variable names.
clear all data = readtable('agaricus-lepiota.data','ReadVariableNames',false);
Display the first 5 mushrooms with their first few features.
data(1:5,1:10)
ans =
Var1 Var2 Var3 Var4 Var5 Var6 Var7 Var8 Var9 Var10
____ ____ ____ ____ ____ ____ ____ ____ ____ _____
'p' 'x' 's' 'n' 't' 'p' 'f' 'c' 'n' 'k'
'e' 'x' 's' 'y' 't' 'a' 'f' 'c' 'b' 'k'
'e' 'b' 's' 'w' 't' 'l' 'f' 'c' 'b' 'n'
'p' 'x' 'y' 'w' 't' 'p' 'f' 'c' 'n' 'n'
'e' 'x' 's' 'g' 'f' 'n' 'f' 'w' 'b' 'k'Extract the first column, labeled data for edible and poisonous groups. Then delete the column.
labels = data(:,1);
labels = categorical(labels{:,:});
data(:,1) = [];Store the names of predictors (features), which are described in agaricus-lepiota.names.
VarNames = {'cap_shape' 'cap_surface' 'cap_color' 'bruises' 'odor' ...
'gill_attachment' 'gill_spacing' 'gill_size' 'gill_color' ...
'stalk_shape' 'stalk_root' 'stalk_surface_above_ring' ...
'stalk_surface_below_ring' 'stalk_color_above_ring' ...
'stalk_color_below_ring' 'veil_type' 'veil_color' 'ring_number' ....
'ring_type' 'spore_print_color' 'population' 'habitat'};Set the variable names.
data.Properties.VariableNames = VarNames;
There are a total of 2480 missing values denoted as '?'.
sum(char(data{:,:}) == '?')ans =
2480Based on the inspection of the data set and its description,
the missing values belong only to the 11th variable (stalk_root).
Remove the column from the table.
data(:,11) = [];
kmedoids only accepts numeric data.
You need to cast the categories you have into numeric type. The distance
function you will use to define the dissimilarity of the data will
be based on the double representation of the categorical data.
cats = categorical(data{:,:});
data = double(cats);kmedoids can use any distance metric
supported by pdist2 to cluster. For this example you will cluster
the data using the Hamming distance because this is an appropriate
distance metric for categorical data as illustrated below. The Hamming
distance between two vectors is the percentage of the vector components
that differ. For instance, consider these two vectors.
v1 = [1 0 2 1];
v2 = [1 1 2 1];
They are equal in the 1st, 3rd and 4th coordinate. Since 1 of the 4 coordinates differ, the Hamming distance between these two vectors is .25.
You can use the function pdist2 to measure
the Hamming distance between the first and second row of data, the
numerical representation of the categorical mushroom data. The value
.2857 means that 6 of the 21 features of the mushroom differ.
pdist2(data(1,:),data(2,:),'hamming')ans =
0.2857In this example, you’re clustering the mushroom
data into two clusters based on features to see if the clustering
corresponds to edibility. The kmedoids function
is guaranteed to converge to a local minima of the clustering criterion;
however, this may not be a global minimum for the problem. It is a
good idea to cluster the problem a few times using the 'replicates' parameter.
When 'replicates' is set to a value, n,
greater than 1, the k-medoids algorithm is run n times,
and the best result is returned.
To run kmedoids to cluster data into 2
clusters, based on the Hamming distance and to return the best result
of 3 replicates, you run the following.
rng('default'); % For reproducibility [IDX, C, SUMD, D, MIDX, INFO] = kmedoids(data,2,'distance','hamming','replicates',3);
Let's assume that mushrooms in the predicted group 1 are poisonous and group 2 are all edible. To determine the performance of clustering results, calculate how many mushrooms in group 1 are indeed poisonous and group 2 are edible based on the known labels. In other words, calculate the number of false positives, false negatives, as well as true positives and true negatives.
Construct a confusion matrix (or matching matrix), where the
diagonal elements represent the number of true positives and true
negatives, respectively. The off-diagonal elements represent false
negatives and false positives, respectively. For convenience, use
the confusionmat function, which calculates a
confusion matrix given known labels and predicted labels. Get the
predicted label information from the IDX variable. IDX contains
values of 1 and 2 for each data point, representing poisonous and
edible groups, respectively.
predLabels = labels; % Initialize a vector for predicted labels. predLabels(IDX==1) = categorical({'p'}); % Assign group 1 to be poisonous. predLabels(IDX==2) = categorical({'e'}); % Assign group 2 to be edible. confMatrix = confusionmat(labels,predLabels)
confMatrix =
4176 32
816 3100Out of 4208 edible mushrooms, 4176 were correctly predicted to be in group 2 (edible group), and 32 were incorrectly predicted to be in group 1 (poisonous group). Similarly, out of 3916 poisonous mushrooms, 3100 were correctly predicted to be in group 1 (poisonous group), and 816 were incorrectly predicted to be in group 2 (edible group).
Given this confusion matrix, calculate the accuracy, which is the proportion of true results (both true positives and true negatives) against the overall data, and precision, which is the proportion of the true positives against all the positive results (true positives and false positives).
accuracy = (confMatrix(1,1)+confMatrix(2,2))/(sum(sum(confMatrix)))
accuracy =
0.8956precision = confMatrix(1,1) / (confMatrix(1,1)+confMatrix(2,1))
precision =
0.8365The results indicated that applying the k-medoids algorithm to the categorical features of mushrooms resulted in clusters that were associated with edibility.
Input Arguments
Name-Value Arguments
Output Arguments
More About
References
[1] Kaufman, L., and Rousseeuw, P. J. (2009). Finding Groups in Data: An Introduction to Cluster Analysis. Hoboken, New Jersey: John Wiley & Sons, Inc.
[2] Park, H-S, and Jun, C-H. (2009). A simple and fast algorithm for K-medoids clustering. Expert Systems with Applications. 36, 3336-3341.
[3] Schlimmer,J.S. (1987). Concept Acquisition Through Representational Adjustment (Technical Report 87-19). Doctoral dissertation, Department of Information and Computer Science, University of California, Irvine.
[4] Iba,W., Wogulis,J., and Langley,P. (1988). Trading off Simplicity and Coverage in Incremental Concept Learning. In Proceedings of the 5th International Conference on Machine Learning, 73-79. Ann Arbor, Michigan: Morgan Kaufmann.
[5] Duch W, A.R., and Grabczewski, K. (1996) Extraction of logical rules from training data using backpropagation networks. Proc. of the 1st Online Workshop on Soft Computing, 19-30, pp. 25-30.
[6] Duch, W., Adamczak, R., Grabczewski, K., Ishikawa, M., and Ueda, H. (1997). Extraction of crisp logical rules using constrained backpropagation networks - comparison of two new approaches. Proc. of the European Symposium on Artificial Neural Networks (ESANN'97), Bruge, Belgium 16-18.
[7] Bache, K. and Lichman, M. (2013). UCI Machine Learning Repository [http://archive.ics.uci.edu/ml]. Irvine, CA: University of California, School of Information and Computer Science.
Extended Capabilities
Version History
Introduced in R2014b
See Also
clusterdata | kmeans | linkage | silhouette | pdist | linkage | evalclusters