mspalign
Align mass spectra from multiple peak lists from LC/MS or GC/MS data set
Description
[ aligns mass spectra from multiple peak
lists (centroided data), by first estimating CMZ,AlignedPeaks]
= mspalign(Peaklist)CMZ, a vector of common
mass/charge (m/z) values estimated by considering the peaks in all spectra in
Peaklist, a cell array of peak lists, where each element corresponds
to a spectrum or retention time. It then aligns the peaks in each spectrum to the values in
CMZ, creating AlignedPeaks, a cell array of
aligned peak lists.
[calls
CMZ, AlignedPeaks] = mspalign(Peaklist,Name,Value)mspalign uses additional options specified by one or more
Name,Value pair arguments.
Examples
Load a MAT-file, which contains liquid chromatography/mass spectrometry (LC/MS) data variables, including peaks and ret_time. peaks is a cell array of peak lists, where each element is a two-column matrix of m/z values and ion intensity values, and each element corresponds to a spectrum or retention time. ret_time is a column vector of retention times associated with the LC/MS data set.
load lcmsdataResample the unaligned data.
[MZ,Y] = msppresample(ms_peaks,5000);
Display the data in a heat map.
msheatmap(MZ,ret_time,log(Y));
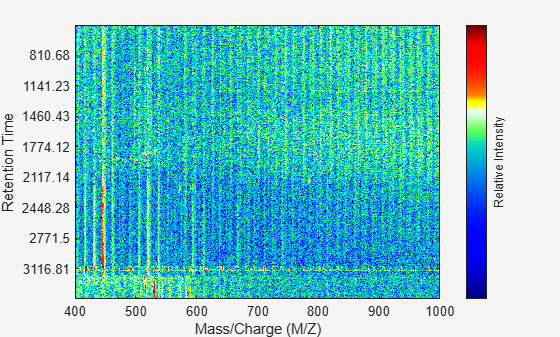
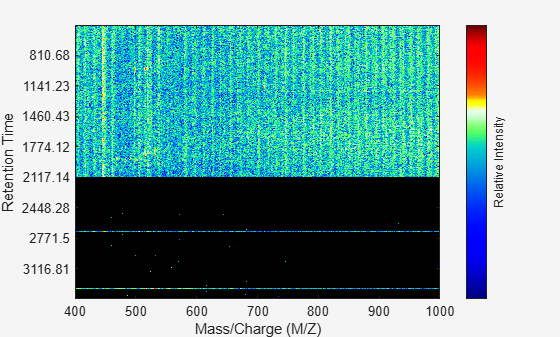
Overlay a dot plot. Zoom in to see how the dots representing peaks overlay the heatmap image.
msdotplot(ms_peaks,ret_time);
Align the peak lists from the mass spectra using the default estimation and correction methods.
[CMZ, aligned_peaks] = mspalign(ms_peaks);
Resample the unaligned data.
[MZ2,Y2] = msppresample(aligned_peaks,5000);
Display it in a heat map.
msheatmap(MZ2,ret_time,log(Y2));
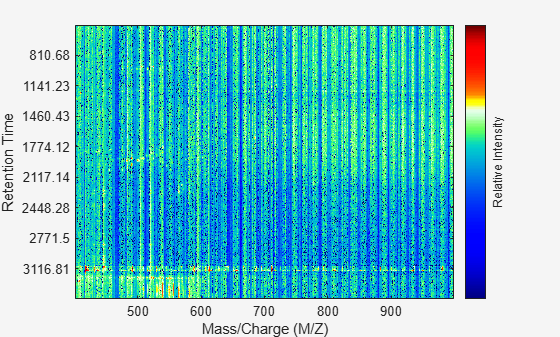
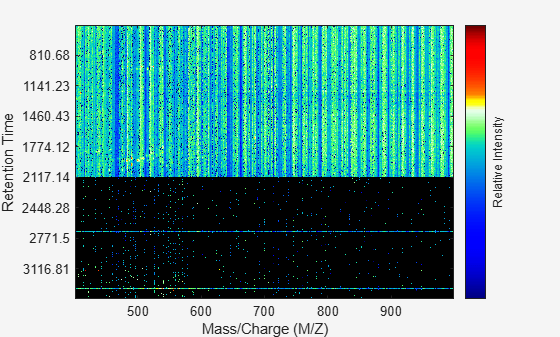
Overlay a dot plot.
msdotplot(aligned_peaks,ret_time);
Input Arguments
Name-Value Arguments
Output Arguments
References
[1] Jeffries, N. (2005) Algorithms for alignment of mass spectrometry proteomic data. Bioinfomatics 21:14, 3066–3073.
[2] Purvine, S., Kolker, N., and Kolker, E. (2004) Spectral Quality Assessment for High-Throughput Tandem Mass Spectrometry Proteomics. OMICS: A Journal of Integrative Biology 8:3, 255–265.
Version History
Introduced in R2007a
See Also
msbackadj | msdotplot | msalign | msheatmap | mslowess | msnorm | mspeaks | msresample | msppresample | mssgolay | msviewer
Topics
- Mass Spectrometry and Bioanalytics
- Preprocessing Raw Mass Spectrometry Data
- Visualizing and Preprocessing Hyphenated Mass Spectrometry Data Sets for Metabolite and Protein/Peptide Profiling
- Differential Analysis of Complex Protein and Metabolite Mixtures Using Liquid Chromatography/Mass Spectrometry (LC/MS)