msppresample
Resample signal with peaks while preserving peaks
Syntax
[X, Intensities]
= msppresample(Peaklist, N)
msppresample(Peaklist, N,
...'Range', RangeValue, ...)
msppresample(Peaklist, N,
...'FWHH', FWHHValue,
...)
msppresample(Peaklist, N,
...'ShowPlot', ShowPlotValue, ...)
Input Arguments
Peaklist | Either of the following:
Tip You can use the |
N | Integer specifying the number of equally spaced points (separation-unit values) in the resampled signal. |
RangeValue | 1-by-2 vector specifying the minimum
and maximum separation-unit values for the output matrix Intensities. RangeValue must
be within [min(,
where inputSU is the concatenated separation-unit
values from the input Peaklist. Default
is the full range [min(. |
FWHHValue | Value that specifies the full width at
half height (FWHH) in separation units. The FWHH is used to convert
each peak to a Gaussian shaped curve. Default is median(diff(,
where inputSU is the concatenated separation-unit
values from the input Peaklist. The default
is a rough approximation of resolution observed in the input data, Peaklist.Tip To ensure that the resolution of the peaks is preserved, set |
ShowPlotValue | Controls the display of a plot of an
original and resampled signal. Choices are true, false,
or I, an integer specifying the index of
a signal in Intensities. If you set to true,
the first signal in Intensities is plotted.
Default is:
|
Output Arguments
X | Vector of equally spaced, common separation-unit values for
a set of signals with peaks. The number of elements in the vector
equals N, or the number of rows in matrix Intensities. |
Intensities | Matrix of reconstructed intensity values for a set of peaks
that share the same separation-unit range. Each row corresponds to
a separation-unit value, and each column corresponds to either a set
of signals with peaks or a retention time. The number of rows equals N,
or the number of elements in vector X. |
Description
Tip
Use the following syntaxes with data from any separation technique that produces signal data, such as spectroscopy, NMR, electrophoresis, chromatography, or mass spectrometry.
[ resamples X, Intensities]
= msppresample(Peaklist, N)Peaklist,
a peak list, by converting centroided peaks to a semicontinuous, raw
signal that preserves peak information. The resampled signal has N equally
spaced points. Output X is a vector of N elements
specifying the equally spaced, common separation-unit values for the
set of signals with peaks. Output Intensities is
a matrix of reconstructed intensity values for a set of peaks that
share the same separation-unit range. Each row corresponds to a separation-unit
value, and each column corresponds to either a set of signals with
peaks or a retention time. The number of rows equals N.
msppresample uses a Gaussian kernel to reconstruct
the signal. The intensity at any given separation-unit value is taken
from the maximum intensity of any contributing (overlapping) peaks.
Tip
msppresample is useful to prepare a set of
signals for imaging functions such as msheatmap and
preprocessing functions such as msbackadj and msnorm.
msppresample( calls Peaklist, N,
... 'PropertyName', PropertyValue, ...)msppresample with
optional properties that use property name/property value pairs. You
can specify one or more properties in any order. Each PropertyName must
be enclosed in single quotation marks and is case insensitive. These
property name/property value pairs are as follows:
msppresample( specifies
a separation-unit range for the output matrix Peaklist, N,
...'Range', RangeValue, ...)Intensities using
the minimum and maximum separation values specified in the 1-by-2
vector RangeValue. RangeValue must
be within [min(,
where inputSU) max(inputSU)]inputSU is the concatenated separation-unit
values from the input Peaklist. Default
is the full range [min(inputSU)
max(inputSU)]
msppresample( sets the full width at half height (FWHH) in separation
units. The FWHH is used to convert each peak to a Gaussian shaped
curve. Default is Peaklist, N,
...'FWHH', FWHHValue,
...)median(diff(,
where inputSU))/2inputSU is the concatenated separation-unit
values from the input Peaklist. The default
is a rough approximation of resolution observed in the input data, Peaklist.
Tip
To ensure that the resolution of the peaks is preserved, set FWHHValue to
half the distance between the two peaks of interest that are closest
to each other.
msppresample( controls the display of a plot of an original
and resampled signal. Choices are Peaklist, N,
...'ShowPlot', ShowPlotValue, ...)true, false,
or I, an integer specifying the index of
a signal in Intensities. If you set to true,
the first signal in Intensities is plotted.
Default is:
false— When return values are specified.true— When return values are not specified.
Examples
Load a MAT-file, included with the Bioinformatics Toolbox™ software, that contains liquid chromatography/mass spectrometry (LC/MS) data variables. It includes
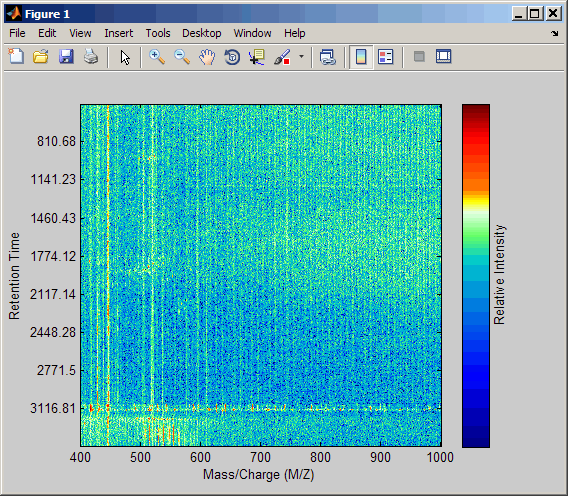
peaks, a cell array of peak lists, where each element is a two-column matrix of m/z values and ion intensity values, and each element corresponds to a spectrum or retention time.load lcmsdataResample the data, specifying 5000 m/z values in the resampled signal. Then create a heat map of the LC/MS data.
[MZ,Y] = msppresample(ms_peaks,5000); msheatmap(MZ,ret_time,log(Y))
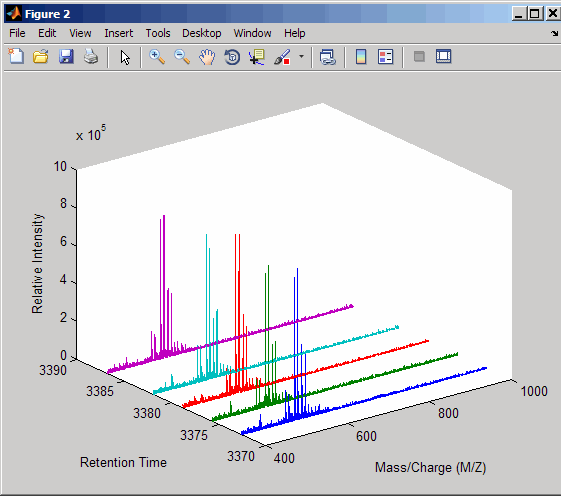
Plot the reconstructed profile spectra between two retention times.
figure t1 = 3370; t2 = 3390; h = find(ret_time>t1 & ret_time<t2); [MZ,Y] = msppresample(ms_peaks(h),10000); plot3(repmat(MZ,1,numel(h)),repmat(ret_time(h)',10000,1),Y) xlabel('Mass/Charge (M/Z)') ylabel('Retention Time') zlabel('Relative Intensity')
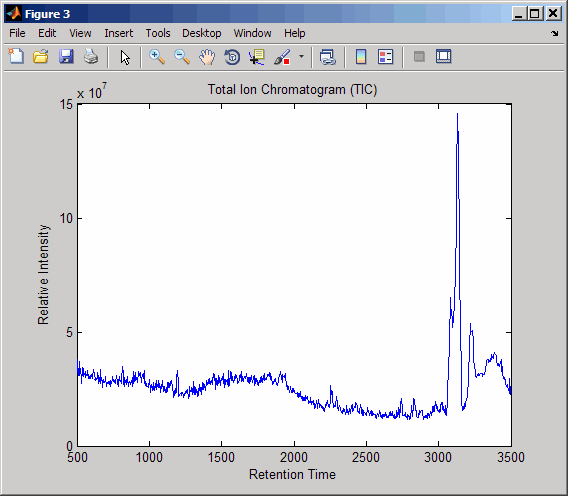
Resample the data to plot the Total Ion Chromatogram (TIC).
figure [MZ,Y] = msppresample(ms_peaks,5000); plot(ret_time,sum(Y)) title('Total Ion Chromatogram (TIC)') xlabel('Retention Time') ylabel('Relative Intensity')
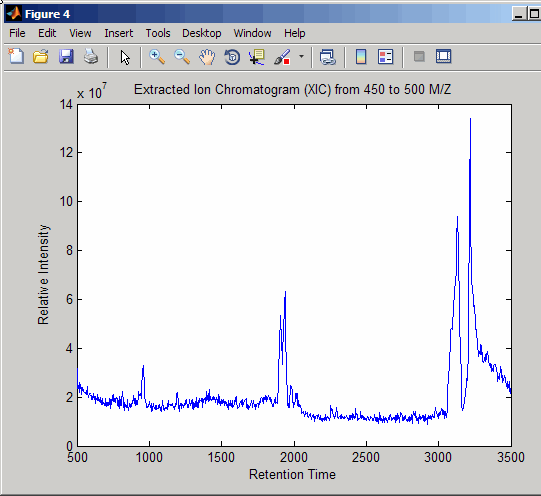
Resample the data to plot the Extracted Ion Chromatogram (XIC) in the 450 to 500 m/z range.
figure [MZ,Y] = msppresample(ms_peaks,5000,'Range',[450 500]); plot(ret_time,sum(Y)) title('Extracted Ion Chromatogram (XIC) from 450 to 500 M/Z') xlabel('Retention Time') ylabel('Relative Intensity')
Version History
Introduced in R2007a
See Also
mspalign | msbackadj | msdotplot | msalign | msheatmap | mslowess | msnorm | mspeaks | msresample | mssgolay | msviewer
Topics
- Mass Spectrometry and Bioanalytics
- Preprocessing Raw Mass Spectrometry Data
- Visualizing and Preprocessing Hyphenated Mass Spectrometry Data Sets for Metabolite and Protein/Peptide Profiling
- Differential Analysis of Complex Protein and Metabolite Mixtures Using Liquid Chromatography/Mass Spectrometry (LC/MS)